The toxicity of azidothymidine (AZT) on human and animal cells in culture at concentrations used for antiviral therapy
Genetica 95: 103-109,1995.
©1995 Kluwer Academic Publishers. Printed in the Netherlands.
Received 10 October 1994 Accepted 11 November 1994
Key words: chemotherapy, discrepancies in cytotoxic dose, AZT-resistant cell variants
Abstract
AZT, a chain terminator of DNA synthesis originally developed for chemotherapy, is now prescribed as an antihuman immunodeficiency virus (HIV) drug at 500 to 1500 mg/person/day, which corresponds to 20 to 60 µM AZT. The human dosage is based on a study by the manufacturer of the drug and their collaborators, which reported in 1986 that the inhibitory dose for HIV replication was 0.05 to 0.5 µM AZT and that for human T-cells was 2000 to 20,000 times higher, i.e. 1000 ~M AZT. This suggested that HIV could be safely inhibited in humans at 20 to 60 µM AZT. However, after the licensing of AZT as an anti-HIV drug, several independent studies reported 20to 1000-fold lower inhibitory doses of AZT for human and animal cells than did the manufacturer's study, ranging from 1 to 50 µM. In accord with this, life threatening toxic effects were reportod in humans treated with AZT at 20 to 60 µM. Therefore, we have re-examined the growth inhibitory doses of AZT for the human CEM T-cell line and several other human and animal cells. It was found that at 10 µM and 25 µM AZT, all cells are inhibited at least 50% after 6 to 12 days, and between 20 and 100% after 38 to 48 days. Unexpectedly, variants of all cell types emerged over time that were partially resistant to AZT. It is concluded that AZT, at the dosage prescribed as an anti-HIV drug, is highly toxic to human cells.
Introduction
AZT (3'-azido-3'-deoxythymidine) is an analog of thymidine in which the 3' hydroxyl group is replaced by an azido group. This prevents the extension of a growing DNA strand ending with AZT to the five prime end of another nucleotide triphosphate. Thus AZT functions as a chain terminator of DNA synthesis.
AZT was originally designed in the 1960s to be used as chemotherapy for leukemia (Horwitz, Chua & Noel, 1964). The rationale for cancer chemotherapy is to kill cancer cells during mitosis with cytotoxic chemicals like AZT. Because chemicals cannot distinguish cancer cells from normal cells, the price for chemotherapy is the death of normal cells that are in mitosis. Therefore, chemotherapy must be restrictod to days or weeks. Successful chemotherapy kills the cancer before it kills the host.
Since 1987, chronic administration of AZT and similar nucleoside analogs, like ddC and ddI, have been prescribed to AIDS patients to inhibit human immunodeficiency virus (HIV), the presumed cause of AIDS (Fischl et al., 1987; Richman et al., 1987; Yarchoan et al., 1991). Since 1990, AZT has also been prescribed to healthy HIV antibody-positive persons to prevent AIDS (Volberding et al., 1990; Tokars et al., 1993; Seligmann et al., 1994). The rationale is to inhibit HIV DNA synthesis at doses that do not inhibit cell DNA synthesis (Yarchoan et al., 1991). It is claimed by Burroughs-Wellcome, the manufacturer of AZT, and its collaborators that this can be achieved, because AZT would inhibit DNA synthesis with HIV DNA polymerase in vitro 100 times more effectively than DNA synthesis with cellular DNA polymerase (Furman et al., 1986). Moreover, this study claimed that in vivo AZT was 2000 to 20,000 times more inhibitory to HIV replication, i.e. at 0.05 to 0.5 /1M, than to cell division, i.e. at 1000 µM (Furman et al., 1986).
Accordingly, anti-HIV doses of AZT were chosen to fall into this therapeutic window, e.g. to be 500 to 1500 mg per person per day, or about 20 to 60 µM per kg per day (Furman et al., 1986; Fischl et al., 1987; Volberding et al., 1990; Physicians' Desk Reference, 1994).
However, in view of its inherent cytotoxicity, AZT has been questioned as an acceptable anti-HIV drug on three theoretical grounds (Duesberg, 1992):
(i) Even if AZT were to inhibit HIV DNA synthesis 100 times more than cell DNA synthesis, it could not 'selectively' inhibit HIV, as is claimed by the manufacturer (Furman et al., 1986). Since HIV DNA measures only 10 kb and cell DNA measures 106 kb, and since both DNAs are made in vivo simultancously inside the same cell, cell DNA provides a 105-fold bigger DNA target for AZT toxicity than does HIV DNA. Therefore, the 100-fold higher selectivity of AZT claimed for HIV DNA synthesis is immaterial.
(ii) Inhibition of HIV DNA synthesis in HIVantibody positive persons is completely unnecessary, because HIV does not spread in tte presence of antiviral antibody (Duesberg, 1992). It is for this reason that only about 1 in 1000 T-cells is ever infected in HIV-antibody positive persons (Duesberg, 1992). The fact that only about 0.1% of all susceptible T-cells are ever infected by HIV in HIV-positive persons with and without AIDS proves that HIV is very effectively neutralized by antiviral immunity. Moreover, there is no correlation between the number of HIV-infected cells and AIDS (Duesberg, 1993; Piatak et al., 1993). For example, there are healthy HIV-positive persons who have 30 to 40 times more HIV-infected cells than AIDS patients (Simmonds et al., 1990; Bagasra et al., 1992; Duesberg, 1992).
(iii) Since only about 1 in lOOO T-cells are ever infected by HIV in persons with or without AIDS (Duesberg, 1992; 1992), AZT must kill 999 uninfected cells in order to kill just one HIV-infected cell - a very poor pharmacological index.
Thus theory predicts that AZT cannot selectively restrict HIV replication in vivo. AZT can only inhibit HIV by killing infectod and uninfected target cells. Theory further predicts that AZT is unacceptable as anti-HIV therapy in HIV-antibody positive persons, because it will kill 999 uninfected cells for every infected cell.
In response to these theoretical considerations it is argued by the manufacturer of AZT and its collaborators that, contrary to expectations, AZT is an effective anti-HIV drug, because cell division was observed to be 2000 to 20,000 times more drug-resistant than HIV replication (Furman et al., 1986).
However, after AZT had been licensed for human use, several independent studies reported that the drug is about 20 to 1000 times more toxic to human cells in culture than the manufacturer had claimed, i.e. that the half inhibitory doses (ID 50) ranged between 1 and 50 µM (Table 1). In accordance with these results, life threatening toxicity including anemia, leukopenia, nausea, muscle atrophy, dementia, hepatitis and mortality, has been documented in humans treated with 20 to 60 µM AZT (Mir & Costello, 1988; Duesberg, 1992; Freiman et al., 1993; Tokars et al., 1993; Bacellar et al., 1994; Goodert et al., 1994; Seligmann et al., 1994). If these results were correct, both the dosage of AZT prescribed to humans and the advisability of AZT as an anti-HIV drug need to be reconsidered.
In view of up to a 1000-fold discrepancy between the cytotoxicity of AZT reported by the manufacturer and his collaborators (Furman et al., 1986) and the cytotoxicitiesreported by other investigators (Table 1), we set out to redetermine the cytotoxicity of AZT. We have investigated the effects of AZT on the human CEM T-cell line and on several other human and animal cells in culture. In contrast to the previous studies, that measured toxicity over 1 to 3 rounds of mitoses, we decided to measure long-term toxicity over several weeks, representing up to 24 consecutive cell divisions. We reasoned that this experimental design would more closely mimic human exposure, which is indefinite, extending over numerous mitoses (Fischl et al., 1987; Volberding et al., 1990; Physicians' Desk Reference, 1994). Under the conditions AZT is prescribed as an anti-HIV drug, i.e. chronic application, it could indeed be more toxic than it is after only one or a few mitoses studied earlier, because non-lethal mutations would accumulate in surviving cells. Our experimental design would detect cumulative mutational toxicity acquired over several mitoses, in addition to the complete cytotoxicity observed in one or a few mitoses.
Materials and methods
Materials. RPMI 1640 medium, Dulbecco's Modified Eaglets medium, and fetal bovine serum were purchased from Gibco Laboratories (Grand Island, NY). Serum Plus was purchased from JRH Biosciences (Lenexa, KS). AZT was purchased from Sigma Chemical Co. (St. Louis, MO).
Table 1. 50% inhibitory dose of AZT for human and animal cells as reported by various laboratories.
|
Study
|
Cell type
|
ID50 at µM AZT
|
|
(Furman et al., 1986) |
human T-cell, line H9 |
1000 |
|
(Balzarini, Herdewijn & De Clercq, 1989) |
human T-cell, line CEM |
> 1000 |
|
(Mansuri et al., 1990) |
human T-cell, line CEM |
54 |
|
(Lemaître et al., 1990) |
humanT-cell, line CEM |
36 |
|
(Avramis et al., 1989) |
human T-cell, line CEM |
4 |
|
(Sommadossi et al., 1990) |
human bone marrow |
1 |
|
|
human bone marrow |
5 |
|
(Inoue et al., 1989) |
human bone marrow |
5 |
|
|
human bone marrow |
25 |
|
(Mansuri et al., 1990) |
mouse bone marow |
1.5 |
|
(Gogu, Beckman & Agrawal, 1989) |
mouse bone marrow |
2 |
|
|
mouse fetal liver |
1 |
Culture conditions of cells grown in suspension. The CEM human Iymphoid T-cell line was provided by Robert F. Garry, Tulane University School of Medicine, New Orleans, LA. CEM T-cells were suspended in 5 mL of RPMI 1640 medium enriched with 10% Serum Plus in tissue culture flasks (25 cm2 growth area, Falcon) and were propagated at 37°C in humidified air with 6.5% CO2. The CEM T-cells were maintained at a density around 3 x 105 cells per mL by diluting them 1:2 every other day. The medium was changed every day by spinning down the cells for 5 min in a clinical centrifuge at 6000 rpm and then resuspending them in fresh medium. AZT was added twice every day at 10 and 25 µM concentrations by micropipets with sterile tips. AZT additions were made at about 12 h intervals. A control flask of CEM T-cells was passaged identically without the addition of AZT. A 10 /1L aliquot of evenly distributed cells was counted every other day with a hematocytometer.
Culture conditions of cells grown attached to Petri dishes. The C3H mouse fibroblast cell line, the Hs-27 human foreskin cell line and the WI-38 human lung cell line were purchased from the American Type Culture Collection. The secondary Chinese Hamster lung cells were prepared from animals in our lab. Each of these cell types was cultured while attached to Petri dishes (100 x 20 mm, Falcon) in 10 mL of Dulbeccots Mod ified Eagle's medium enriched with 10% fetal bovine serum at 37°C in humidified air with 6.5% CO2. Each of the monolayer cell types was sceded of approximateIy I X 105 cells on a 10-cm dish containing 10 mL of medium. The medium in each dish was changed every day. AZT additions were also made twice a day at 10 and 25 µM concentrations. The cells were countod with a Coulter counter by placing a 200 pL sample of evenly distributed cells in 10 mL of isotonic buffered saline solution. Each AZT-treated culture was split 1:5 when the control dish had reached 100% confluency.
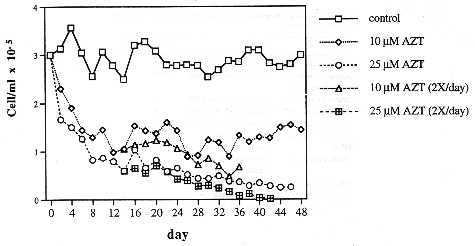
Fig. 1. The effect of AZT, at 10 µM and 25 µM, on the growth rate of the human CEM T-cell line maintained as described in the text.
Results
The effect of long-term AZT treatment on the viability of the human CEM T-cell line. To determine the cytotoxicity of AZT on the human CEM T-cell line in culture, parallel cultures were incubated with 10 µM, 25 µM AZT and without AZT (see Materials and methods). The untreated cells were maintained at saturation density of CEM cells, which is about 3 x 105 cells per mL in our conditions. Each culture was divided 2-fold every 48 h, by which time the AZT-free control had regained saturation density.
As can be seen in Fig. 1, after four days the cell count of the culture at 25 µM AZT had been reduced to half of the control, and that of the culture at 10 µM AZT to two thirds of the control. After 12 days the cell densities of both AZT-treated cultures had been reduced to a third of the control culture. From then on, the density of the culture at 25 µM AZT continued to decline at a decreasing rate, and that of the culture at 10 µM AZT stabilized (Fig. 1).
One possible explanation of the decreasing sensitivity of surviving CEM cells to AZT over time is that the dividing portion of the cells takes up all AZT in a short time, and that the resting portion of cells subsequently enters mitosis in a culture depletod of AZT. Another explanation suggests that variants are selected that do not incorporate AZT into DNA. To distinguish between these possibilities each AZT-treated culture was further divided into two. One of the two subcultures was maintained with daily medium changes containing 10 and 25 µM AZT respectively as before. The other subculture was supplemented, 12 hours after the medium including AZT had been changed, with the equivalent of an extra 10 and 25 µM AZT respectively. All cultures were further incubated under these conditions for another 32 to 36 days.
It can be seen in Fig. I that even at two daily applications of AZT at 10 µM, a decreasing fraction of T-cells retained viability for 14 days (when the culture became contaminated). However, no survivors were observed after 14 days at two daily applications of 25 µM AZT.
It is concluded that T-cell variants are selected, on long-term exposure to AZT, that are relatively resistant to AZT compared to the average T-cell prior to treatment.
The effect of long-term exposure to AZT on the viability of human and animal fibroblasts. To determine whether other human and animal cells are similar to human T-cells with regard to AZT-sensitivity, the viability of a human lung (WI) and foreskin (Hs) cell line, of a mouse cell line (C3H) and of secondary Chinese hamster cells (C.H.) was studied in AZT.
Each of the different cell types was seeded at 1 x 105 cells per 10 cm dish and exposed to AZT at 10 and 25 µM (see Materials and methods). AZT was added to each dish twice every day, once in the morning and again at night as described above. The inhibition of cell growth was expressed as the percentage of cells in the AZT culture compared to that of the untreated control. The cells were counted by the time the control had reached confluency (Fig. 2). The first count of cells was taken at the end of two weeks when all control dishes had become completely confluent. Thereafter control cells were split 1:4 and allowed to reach confluency again. This process was repeated several times as shown in Fig. 2.
As can be seen in Fig. 2, the general pattern of AZT-sensitivity observed with T-cells was confirmed with other human and animal cells. C3H mouse cells appeared to be most sensitive to the effects of AZT. At day 14, the densities of C3H cells, maintained at both concentrations of AZT, had already declined to below 50 percent of the control. Possibly due to a counting error, the density of C3H cells at 10 µM AZT appeared lower than that of cells at 25 µM AZT. After the same time, the concentrations of Hs-27, WI-38, and C.H. cells ranged from 50 to 60 percent of the control at 25 µM AZT, and from 60 to 70 percent of the control at 10 µM AZT.
From 14 to 38 days of AZT treatment all fibroblast cells remained at about half the density of the controls. However, the densities of C3H and Hs-27 cell lines gradually increased over time at both concentrations of AZT. By day 38, the density of Hs-27 cells at both AZT concentrations had reached up to 80 percent of the control.
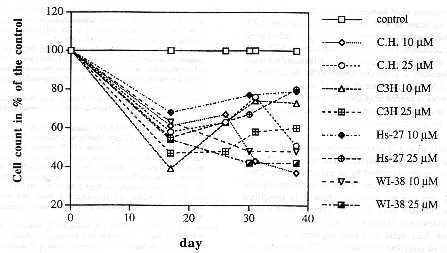
Fig. 2. The effect of AZT, at 10 µM and 25 µM, on the growth of
human lung (Wl), and foreskin cells (Hs), on mouse fibroblasts (C3H)
and on secondary Chinese hamster (C.H.) fibroblasts. AZT-treated
cells were counted whenever the untreated control culture had reached
confluency.
Discussion
(i) AZT toxic to human cells in the micromolar range. Our results indicate that long-term exposure to AZT inhibits the growth of human CEM T-cells about 50% at 10 µM, and gradually up to 100% at 25 µM. Similar results were obtained with human lung and foreskin cells, and also with mouse and Chinese hamster cells, although complete inhibition was not observed with any of these cells under our conditions. Thus our results confirm and extend those of others summarized in Table 1, that AZTis toxic to human cells in the micromolar range. Indeed AZT, like all other nucleotide analogs of DNA,is expectod to be toxic in the micromolar range, because the Michaelis constants of authentic nucleotide triphosphates are also in the micromolar range (Kornberg, 1980).
These results are incompatible with the claim of the manufacturer and its collaborators that AZT is only toxic to human cells in the millimolar range. That claim is also hard to reconcile with the manufacturer's own observation that HIV replication is inhibited by AZT at 0.05 to 0.5 µM (Furman et al., 1986). Since (i) HIV and cell DNA are both replicated in vivo inside the same cellular vesicle and at the same time (Rubin & Temin, 1958; Weiss et al., 1985), (ii) retroviral and cellular DNA synthesis depend on the same triphosphatepools, and (iii) retroviral DNAis a 105-fold smaller target for AZT than cell DNA, HIV DNA synthesis cannot be more sensitive to AZT than cell DNA. In fact target theory predicts the opposite.
Thus the preponderance of evidence casts doubts on the claim of the manufacturer of AZT and its collaborators that AZTis only toxic to cells in the millimolar range (Furman et al., 1986).
(ii) Resistance of human and animal cells to longterm exposure to AZT. Unexpectedly, partially AZT resistant variants emerged from human T-cells and all other cells testod on continued exposure to AZT at 10 to 25 µM for 38 to 48 days. These variants did not reach the densities of untreated control cells, but continued to divide in the presence of AZT at various rates. Further work is needed to analyze the basis for the relative AZT-resistance acquired by human and animal cells upon long-term exposure to AZT.
(iii) Toxicity of AZTat micromolar concentrations calls for reoppraisal of its use as an anti-HlV drug. The cell culture results described by us and others predict that AZT is toxic to humans at the 20 to 60-micromolar level, the concentrations at which it is prescribed as an anti-HIV drug. Even though our results show that human and animal cells acquire some resistance against AZT upon long-term exposure, no cell has achieved complete resistance to AZT under the conditions tested. This prediction is confirmed by numerous clinical studies that describe life threatening toxic effects in humans treatod with AZT at 20 to 60 µM (see Introduction). Thus our data and those of others call into question the merits of AZT as an anti-HIV drug, particularly at the doses currently prescribed to humans.
Acknowledgments
We thank Robert F. Garry, Tulane Univ. New Orleans, for the human CEM T-cell line and for generous advice, and Gedge D. Rosson, UC Berkeley, for preliminary results and discussions. This investigation was supportod in part by the Council for Tobacco Research, USA, and private donations from Thomas Boulger (Redondo Beach, Calif., USA), Glenn Braswell (Los Angeles, Calif., USA), Dr. Richard Fischer (Annandale, Va., USA), Dr. Fabio Franchi (Trieste, Italy), and Dr. Peter Paschen (Hamburg, Germany).
References
Avramis, V.l., W. Markson, R.L. Jackson & E.
Gomperts, 1989. Biochemical pharmacology of zidovudine in human
T-lymphoblastoid cells (CEM). AIDS 3: 417-422.
Bacellar, H., A. Munoz, E.N. Miller, B.A. Cohen,
D. Besley, O.A. Seines, J.T. Becker & J.C. McArthur, 1994.
Temporal trends in the incidence of HIV-I -related neurologic
diseases: Multicenter AIDS CohortStudy, 1985-1992.Neurology44:
1892-1900.
Bagasra, O., S.R Hauptman, H.W. Lischner, M. Sachs
& R.J. Pomerantz, 1992. Detection of human immunodeficiency virus
type I provirus in mononuclear cells by in situ polymerase chain
reaction. N. Engl. J. Med.326: 1385-1391.
Balzarini, J., R Herdewijn & E. De Clercq,
1989. Differential pattems of intracellular metabolism of
2',3'-didehydro-2',3'didexoythymidine and
3'-azido-2',3'-dideoxythymidine, two potent anti-human
immunodeficiency vims compounds. J. Biol. Chem.264:6127 6133.
Duesberg, RH., 1992. AIDS acquired by drug
consumption and other noncontagious risk factors. Pharmacology &
Therapeutics 55: 201-277.
Duesberg, RH., 1993. HIV and AIDS. Science 260:
1705.
Fischl, M.A., D.D. Richman, M.H. Grieco, M.S.
Gottlieb, RA. Volberding, O.L. Laskin, J.M. Leedon, J.E. Groopman, D.
Mildvan, R.T. Schooley, G.G. Jackson, D.T. Durack, D. King and the
AZT Collaborative Working Group, 1987. The effficacy of
azidothymidine (AZT) in the treatment of patients with AIDS and
AlDS-related complex. N. Engl. J. Med.317: 185-191.
Freiman, J.R, K.E. Helfert, M.R. Hamrell &
D.S. Stein, 1993. Hepatomegaly with severe steatosis in
HlV-seropositive patients. AIDS 7: 379-385.
Furman, RA., J.A. Fyfe, M. St Clair, K. Weinhold,
J.L. Rideout, G.A. Freeman, S. Nusinoff-Lehrman, D.R Bolognesi, S.
Broder, H. Mitsuya & D.W. Barry, 1986. Phosphorylation of
3'-azido-3'deoxythymidine and selective interaction of the
5'-triphosphate with human immunodeficiency virus reverse
transcriptase. Proc. Natl. Acad. Sci. USA 83: 8333-8337.
Goedert, J.J., A.R. Cohen, C.M. Kessler, S.
Eichinger, S.V. Seremetis, C.S. Rabkin, F.J. Yellin, RS. Rosenberg
& L.M. Aledort,1994. Risks of immunodeficiency, AIDS, and death
related to purity of factor Vlll concentrate. Lancet 344:
791-792.
Gogu, S.R., B.S. Beckman & K.C. Agrawal, 1989.
Anti-HIV drugs: Comparative toxicities in murine fetal liver and bone
marrow erythroid progenitor cells. Life Sci. 45: iii-vii.
Horwitz, J.R, J. Chua & M. Noel, 1964.
Nucleosides. V. The monomesylates of
1-(2'-deoxy-beta-Dlyxofuranosyl)thymidine. J. Org. Chem. 29:
2076.
Inoue, T., K. Tsushita, T. Itoh, M. Ogura, T.
Hotta, M. Saneyoshi, S. Yoshida, H. Saitoh, Y. Tomoda & Y. Nagai,
1989. In vitro bone marrow toxicity of nucleoside analog against
human immunodeficiency virus. Antimicrob. Agents Chemother. 33:
576-579.
Komberg, A., 1980. DNA Replication. Freeman and
Company, San Francisco.
Lema~tre, M., D. Guetard, Y. Henin, L. Montagnier
& A. Zerial, 1990. Protective activity of tetracycline analogs
against the cytopathic effect of the human immunodeficiency viruses
in CEM cells. Res. Virol. 141: 5-16.
Mansuri, M.M., M.J.M. Hitchcock, R.A. Buroker,
C.L. Bregman, 1. Ghazzouli, J.V. Desiderio, J.E. Starrett, R.Z.
Sterzycki & J.C. Martin, 1990. Comparison of in vitro biological
properties and mouse toxicities of three thymidine analogs active
against human immunodeficiency virus. Antimicrob. Agents Chemother.
34: 637-641.
Mir, N. & C. Costello, 1988. Zidovudine and
bone marrow. Lancet ii: 1195-1196.
Physicians' Desk Reference,1994. Retrovir.
Piatak, M., L.C. Saag, S.C. Yang, S.J. Clark, J.C.
Kappes, K.-C. Luk, B.H. Hahn, G.M. Shaw & J.D. Lifson, 1993. High
levels of HIV-I in plasma during all stages of infection determined
by competitivePCR. Science259: 1749-1754.
Richman, D.D., M.A. Fischl, M.H. Grieco, M.S.
Gottlieb, RA. Volberding, O.L. Laskin, J.M. Leedom, J.E. Groopman,
D.
Mildvan, M.S. Hirsch, G.G. Jackson, D.T. Durack,
S. NusinoffLehrman andthe AZTCollaborative Working Group,1987. The
toxicity of azidothymidine (AZT) in the treatment of patients with
AIDS and AlDS-related complex. N. Engl. J. Med. 317: 192-197.
Rubin, H. & H. Temin, 1958. A radiological
study of cell-virus interaction in the Rous sarcoma. Virology 7:
75-91.
Seligmann, M., D. A. Warrell, J.-R Aboulker, C.
Carbon, J.H. Darbyshire, J. Dormont, E. Eschwege, D.J. Girling, D.R.
James, J.-R Levy, RT.A. Peto, D. Schwarz, A.B. Stone, l.V.D. Weller,
R. Withnall, K. Gelmon, E. Lafon, A.M. Swart, V.R. Aber, A.G.
Babiker, S. Lhoro, A.J. Nunn & M. Vray,1994. Concorde: MRC/ANRS
randomised double-blind controHed trial of immediate and deferred
zidovudine in symptom-free HIV infection. Lancet 343: 871-881.
Simmonds, R, R Balfe, J.F. Peutherer, C.A. Ludlam,
J.O. Bishop & A.J. Leigh-Brown, 1990. Human immunodeficiency
virus-infected individuals contain provirus in small numbers of
peripheral mononuclear cells and at low copy numbers. J. Virol. 64:
864-872.
Sommadossi, J.-R, Z. Zhu, R. Carlisie, M.-Y. Xie,
D.A. Weidner & M.H. El Kouni, l 990. Novel pharmacological
approaches to the treatment of AIDS and potential use of uridine
phosphorylase inhibitors. In: Advances in Chemotherapy of AIDS,
pp.63-73,
R.B. Diasio & J.-R Sommadossi (eds.) Pergamon
Press Inc., New York.
Tokars, J.l., R. Marcus, D.H. Culver, C.A.
Schable, RS. McKibbe, C.l. Bandea & D.M. Bell, 1993. Surveillance
of HIV infection and zidovudine use among health care workers
aÎter occupational exposure to HlV-infectod blood. Ann. Intem.
Med. 118: 913-919.
Volberding, RA., S.W. Lagakos, M.A. Koch, C.
Pettinelli, M.W. Myers, D.K. Booth, H.H. Balfour Jr., R.C. Reichman,
J.A. Bartlett, M.S. Hirsch, R.L. Murphy, W.D. Hardy, R. Soeiro, M.A.
Fischl, J.G. Bartlett, T.C. Merigan, N.E. Hyslop, D.D. Richman, F.T.
Valentine, L. Corey and the AIDS Clinical TAal Group of the National
Institute of Allergy and Infectious Disease, 1990. Zidovudine in
asymptomatic human immunodeficiency virus infection: A controlled
trial in persons with fewer than 500 CD4-positive cells per cubic
millimeter. N. Engl. J. Med. 322: 941-949.
Weiss, R., N. Teich, H. Varmus & J. Coffin,
1985. Molecular Biology of RNA Tumor Viruses. Cold Spring Harbor
Press, Cold Spring Harbor, NY.
Yarchoan, R., J.M. Pluda, C.-F. Pemo, H. Mitsuya
& S. Broder, 1991, Anti-retroviral therapy of human
immunodeficiency virus infection: current strategies and challenges
for the future. Blood 78: 859-884.
| RETOUR À AZT | HOME PAGE | CONTACTS | NOS PUBLICATIONS | COMMANDES et DONATIONS |